
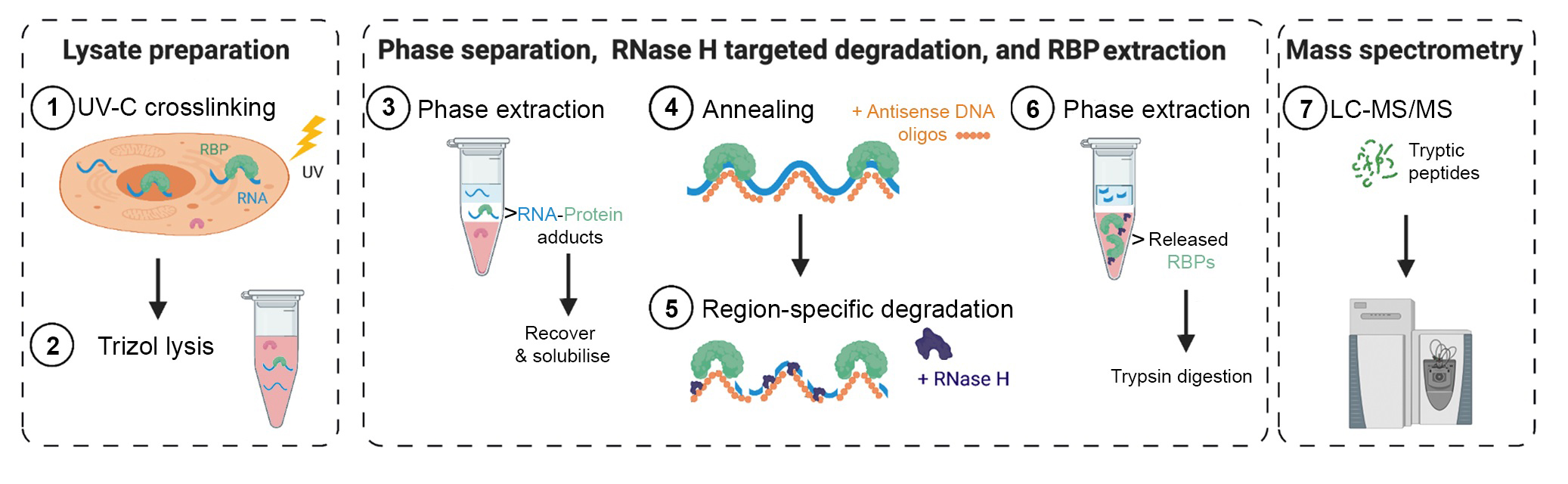
TREX illustrated Step-by-Step Protocol
TREX uses TRIZOL phase separation and interface isolation of
RNA-protein adducts followed by targeted RNA degradation and recovery of released
RNA-binding proteins (RBPs).
Materials
Biological materials
The biological material for our TREX test studies was human HCT116 cells. However, in theory any RNA in any cell that can be UV-C crosslinked and lysed by TRIZOL can be subjected to TREX analysis.
! CAUTION It is important to regularly check cell lines to ensure that they are authentic and are not infected with mycoplasma. Handle cell lines according to the supplier’s instructions. Work in a biosafety hood, use sterile equipment, and wear gloves to minimize the risk of contamination.
Reagents
· RNaseZAP™ Surface Decontaminant (Sigma, R2020)
· Nuclease-free water (Thermo, AM9932)
· Cytosolic Lysis buffer (1% Triton X-100, 10mM Tris-HCl pH 7.5 150 mM NaCl, cOmplete™ protease inhibitor cocktail (Sigma), SUPERase•In™ 1/500 v/v (Invitrogen , AM2694))*
! CAUTION Triton is harmful, and it is an irritant. Triton is hazardous to the environment. Handle solutions containing Triton with care and dispose of waste according to institutional regulations.
! CAUTION cOmplete™ protease inhibitor cocktail is an irritant. Handle solutions containing the protease inhibitor mix with care and dispose of waste according to institutional regulations.
· TRIzol™ LS Reagent (Thermo, 10296028)
· TRIzol™ Reagent (Fisher Scientific Uk Ltd, 12034977)
! CAUTION TRIzol™ reagents are toxic if inhaled and a potential carcinogen. Always handle solutions containing chloroform with care while wearing personal protective equipment in a chemical fume hood and dispose of waste according to institutional regulations.
· Chloroform
! CAUTION Chloroform is volatile and toxic. Chloroform is an irritant. Handle solutions containing chloroform with care and dispose of chloroform waste according to institutional regulations.
· TE (Tris-Cl pH7,10 mM, EDTA 1 mM)*
· TE+SDS 0.1% (Tris-Cl pH7, 10 mM, EDTA 1 mM, SDS 0.1 %)*
· TE+SDS 0.5% (Tris-Cl pH7, 10 mM, EDTA 1 mM, SDS 0.5 %)*
· 1X Hybridization buffer: (50mM NaCl, 1mM EDTA, 100mM TrisHCl pH 7.0) *
· Thermostable RNAse H (NEB, M0523S)
· Thermostable RNAse H buffer (NEB, M0523S)
· Isopropanol
· Proteinase K (Thermo, AM2546)
· Proteinase K buffer (0.1 M NaCl, 10 mM Tris HCl pH 8.0, 1 mM EDTA, 0.5% SDS)*
· Acetone
· Ethanol
· Dithiothreitol (DTT) ≥99.4%, Proteomics Grade (VWR , M109)
· Iodoacetamide (IAA) ≥98%, Proteomics Grade (Sigma, I1149)
· Urea ≥99.5% (Sigma , U1250)
· ABC buffer (50 mM Ammonium Bicarbonate, pH 7.8)
· Trypsin, LC-MS/MS grade (Sigma, 650279)
· Stop buffer (4% acetonitrile, 1% TFA)
· 5M NaCl (Invitrogen REF AM9760G)
· Invitrogen GlycoBlue Coprecipitant 15mg/mL (Fisher Scientific Uk Ltd, 10301575)
· 1M Tris HCl pH 7.0 (Invitrogen, AM9850G)
· 0.5M EDTA pH 8.0 (Invitrogen, AM9260G)
· 1M MgCl2 (Invitrogen, AM9530G)
· 10% SDS (Invitrogen,15553-035)
· TURBO DNase 10X buffer (Fisher Scientific, 10646175)
· TURBO DNase (Fisher Scientific, 10646175)
·
RNasin® Plus RNase inhibitor (Promega, N2611)
* Use RNase-free reagents and Nuclease-free water to prepare these buffers.
Equipment
· Ultraviolet crosslinker (Hoefer, UVC500)
· Cell scrapers (Corning, cat.no 353087)
· 150 mm TC-treated Culture Dish
· RNase-free Microfuge 2 ml tubes (Thermo, AM12425)
· RNase-free Microfuge 1.5 ml tubes
· 0.2 mL 8-Strip PCR tube (Thermo, AM12425)
· Hood for Trizol
· VeritiTM Thermocycler (Thermo, 4375786)
· Thermomixer comfort ( Eppendorf, No. 5355)
· Barrier pipette tips
· Benchtop centrifuge (Eppendord, 5415R)
· C18 Stage tips
· Mass spectrometer
· Vortexer (Scientific industries, SI-0236)
· Qubit 2.0 fluorometer (Invitrogen, Q32866)
· QuantStudio 7 Flex Real-Time PCR (Thermo, 4485701)
· RT-qPCR plates
Software
· Q7 RT-qPCR software
· Maxquant
· Perseus
Previous steps to do before starting:
Seed cells on 150 mm TC-treated Culture Dish and allow attachment for 16 hours in a 37 °C incubator with 5% CO2 prior to crosslinking.
1) In vivo cross-linking. Timing 2 min
1. Aspirate the media and add 15 ml cold PBS.
2. Aspirate the PBS and add another 15 ml of cold PBS.
3. Aspirate the PBS.
4. Transfer the dish onto ice or an ice receptacle that fits into the cross-linker device.
CRITICAL cross-linker device sensor must not be covered to continually measure the UV energy
5. Cross-link cells at 200 mJ/cm2 on ice.
CRITICAL dish lid must be removed from plate to ensure crosslinking efficiency.
Note: The cross-linking energy of 200 mJ/cm2 is optimal for most RNAs we have tested, but this may need further optimisation for certain targets. In particular, lowly abundant RNAs, or very small target regions devoid of many pyrimidines may require higher energy levels for efficient crosslinking.
Note: To perform TREX on total cell RNA proceed with 2A. If prior subcellular fractionation is required, proceed with 2B.
2A) Whole cell lysis. Timing 10 min
6. Add 1ml of TRIzol™ for every 20 million cells on the plate
7. Scrape the cells thoroughly. Make sure the TRIzol™ has covered the whole plate while scraping.
8. Homogenize the lysate through repeated pipetting.
PAUSE POINT Store the lysate for up to 3 months at -80 °C if needed.
2B) Subcellular fractionation. Timing 20 min
9. Scrape cells in 1 mL of RNase-free ice-cold PBS per 20 million cells. Transfer each 1ml of lysate to an 2 ml RNase-free Eppendorf tube.
10. Spin down for 1 minute at 500 g at 4°C to pellet the cells. Discard the PBS supernatant (as shown in the image).
11. Lyse the cell pellet in 250 µl Cytosolic Lysis buffer.
12. Spin for 15 min at 10,000 g at 4°C and collect the supernatant, which contains the cytosolic fraction.
Note: Alternatively keep the remaining pellet if the nuclear fraction is need.
PAUSE POINT Store the fractions for up to 3 months at -80 °C if needed.

3A) TRIzol™ phase separation for whole cell lysate (following on from 2A) Timing 1h
13. Incubate the homogenized lysate for 5 min at room temperature (20–25 °C) to dissociate non-crosslinked RNA-protein interactions.
14. Add 200 µl of chloroform / 1ml of TRIzol™ and vortex sample until the content is homogeneous.
15. Centrifuge for 15 min at 12,000 g at 4°C

CRITICAL Three phases should now be visible (as shown in the image).
16. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without disrupting the interface.
17. Resolubilise the interface in 1ml of TRIzol™ / 20 million cells
18. Add 200 µl of chloroform / 1ml of TRIzol™ and vortex sample until the content is homogeneous
19. Centrifuge for 15 min at 12,000 g at 4°C
20. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without touching the interface.
21. Resolubilise the interface in 1 ml of TRIzol™ / 20 million cells
PAUSE POINT Store the lysate at -80 °C if needed.
22. Add 200 µl of chloroform / 1ml of TRIzol™ and vortex sample until the content is homogeneous
23. Centrifuge for 15 min at 12,000 g at 4°C
24. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without touching the interface.
Note: The following steps are required if you need to work with more than 20 million cells per replicate.
25. Combine the interfaces from up to a 100 million cells in 1ml of TRIzol™ and transfer into a 2 ml tube.
26. Add 200 µl of chloroform and vortex sample until the content is homogeneous.
27. Centrifuge for 15 min at 12,000 g at 4°C.
28. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without touching the interface.
3B) TRIzol™ LS phase separation for subcellular TREX (following on from 2B) Timing 1h
29. Add 0.75 ml of TRIzol™ LS to the sample and homogenize through repeated pipetting.
Note: Use 1 ml of standard TRIzol™ if continuing with the nuclear fraction.
30. Incubate homogenized lysate for 5 min at room temperature (20–25 °C) to dissociate non-crosslinked RNA-protein interactions.
31. Add 200 µl of chloroform and vortex sample until the content is homogeneous.
32. Centrifuge for 15 min at 12,000 g at 4°C.
CRITICAL Three phases should now be visible (as shown in the above image).
33. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without touching the interface.
34. Resolubilise the interface in 1 ml of TRIzol™.
35. Add 200 µl of chloroform and vortex.
36. Centrifuge for 15 min at 12,000 g at 4°C. Remove upper and lower phases as before.
37. Resolubilise the interface in 1 ml of TRIzol™.
PAUSE POINT Store the lysate at -80 °C if needed.
38. Add 200 µl of chloroform and vortex.
39. Centrifuge for 15 min at 12,000 g at 4°C. Remove upper and lower phases as before.
CRITICAL leave no more than a maximum of 50 µl of liquid around the interface.
Note: The following steps are required if you work with more than 20 million cells per replicate.
40. Combine the interface from up to a 100 million cells in 1ml of TRIzol™ and transfer into a 2 ml tube.
41. Add 200 µl of chloroform and vortex sample until the content is homogeneous.
42. Centrifuge for 15 min at 12,000 g at 4°C.
43. Remove the upper aqueous phase until the interface collapses. Remove as much of the lower organic phase as possible without touching the interface.
3C) Interface
solubilisation and DNA removal Timing 4h
Interface wash:
44. Without disturbing the interface, add 1ml of TE buffer
by pipetting it slowly along the wall into the tube.
CRITICAL Don’t resuspend the or
disturb the interface.
45. Slowly invert the tube 3 times, taking extra care not to disturb the interface much.
46. Centrifuge for 1 min at 5000 g at 4°C.
47. Remove the wash supernatant and discard.
48. If needed, repeat step 44 to 47 1 or 2 more times till no more red dye from TRIzol™ is visible.
Note: After centrifugation a small amount of TRIzol™ might appear trapped under the interface pellet. Repeating the wash (step 44-47) will remove this.
Breaking down the interface:
49. Resuspend the interface through pipetting in 1ml of TE+SDS 0.1% for up 30-60 times.
50. When the interface stops to break down any further, centrifuge for 2 min at 5000 g at room temperature.
51. Transfer the supernatant into a new 2ml tube (Collection tube 1)
Note: From this point
onwards, the interface should become increasingly translucent.
CRITICAL Don’t transfer any of the of the interface
into the collection tube.
52. Resuspend the remaining interface through repeated pipetting in 1ml of TE+SDS 0.1% for up 30-60 times.
53. Centrifuge for 2 min at 5000 g at room temperature.
54. Transfer the supernatant into a new 2ml tube (Collection tube 2)
55. Resuspend the remaining interface through repeated pipetting in 1ml of TE+SDS 0.5% for up 30-60 times.
56. Centrifuge for 2 min at 5000 g at room temperature.
57. Transfer the supernatant into a new 2ml tube (Collection tube 3)
58. Resuspend the remaining interface through pipetting in 1ml of TE+SDS 0.5% for up 30-60 times.
59. Centrifuge for 2 min at 5000 g at room temperature.
60. Transfer the supernatant into a new 2ml tube (Collection tube 4)
Precipitation 1:
61. To each collection tube add 60 µl of NaCl 5M, 1 µl Glycoblue and 1ml of isopropanol.
62. Invert the tube multiple times.
PAUSE POINT Samples can be stored at -20 °C overnight if needed.
63. Centrifuge for 15 min at 18000 g (or faster) at 4°C (or colder).
Ethanol wash:
64. Carefully remove the supernatant from all collection tubes.
65. Add 1ml of 70% ethanol to the first collection tube and break down the pellet through repeated pipetting. Then transfer the content to the next collection tube and repeat the process for all 4 tubes until all the pellets are combined in the same Ethanol solution.
66. Use another 1 ml of 70% ethanol to rinse any residual pellets from the now empty collection tubes. Combine this solution with the collection from step 35, leaving you with 2ml of ethanol suspension containing all the pellets.
67. Centrifuge for 1 min at 18000 g at room temperature.
68. Discard the supernatant. Remove any remaining ethanol using an additional short spin.
Hydration:
Note: The following
volumes are for up to 100 million cells.
69. Add 1.8ml of nuclease-free water to the pellet.
70. Invert the tube multiple times or vortex briefly to detach the pellet from the bottom the tube.
71. Let the pellet hydrate for 1 hour on ice with occasional inversions.
Note: Pellet will become translucent.
72. Using a 1 ml pipette, resuspend the pellet repeatedly until it is completely dissolved.
DNase treatment:
73. Add 200 µl of 1X TURBO DNase buffer and mix the solution
through pipetting.
74. Add 2 µl of RNase inhibitor and 100 µl of TURBO DNase.
75. Invert tube 4-5 times and incubate for 50 min at 37 °C, 700 rpm shaking.
Precipitation 2:
76. Split sample into two 2ml tubes (1ml each)
77. To each collection tube add 60 µl of NaCl 5M and 1ml of isopropanol
78. Invert the tube multiple times.
PAUSE POINT Samples can be stored at -20 °C overnight if needed.
79. Centrifuge for 15 min at 18000 g (or faster) at 4°C (or colder).
Ethanol wash 2:
80. Carefully remove the supernatant from each tube.
81. Add 1ml of 70% ethanol to the first tube and break down the pellet through pipetting. Transfer the content to the second tube and repeat the process until the pellets are combined in the same ethanol solution.
82. Use another 1 ml of 70% ethanol to rinse any residual pellets from the now empty tubes. Combine this solution with the collection from step 81, leaving you with 2ml of ethanol containing the combined pellet.
83. Centrifuge for 1 min at 18000 g at room temperature
84. Discard the supernatant and remove any remaining ethanol using an additional short spin.
4) Probe Hybridization (annealing). Timing 1h
85.
Dissolve the pellet through repeated pipetting in 110 µl of Hybridization buffer
per 20 million cells (e.g. if you started with 100 million cells per replicate,
dissolve in 550 µl of hybridization buffer).
CRITICAL Pipette until the pellet does not
dissolve any further.
86. Add 6 µl of 1 – 200 µM of tiling DNA oligos* per 20 million cells**
(e.g.: for U1 snRNA, we used 6 µl of 10 µM oligos per 20 million cells. Note that oligos are added to both control and experimental samples)
87. Invert samples 4-5 times and give them a brief spin.
Summary table for the samples:
|
RNA/PROBE HYBRIDIZATION REACTION |
VOLUME for 20 million cells |
|
Total RNA in Hybridization Buffer |
110 µl |
|
Oligonucleotide pool |
6 µl 1-200 µM |
|
Total Volume |
116 µl |
*TREX uses antisense tiling DNA oligos designed against a target RNA of interest. Oligos must be non-overlapping and provide a complete coverage of the target sequence of interest. Our oligos are on average 60 nt in length (but not smaller than 30 or longer than 90). We ensure that no oligos strongly match to an off-target region by blasting the sequences against the RefSeq database.
** The amount of cells and oligos to use will depend on each target RNA and its copy number in your cells and must be empirically determined through qPCR analysis of target depletion for each set of oligos.
88. The annealing will be performed in a Thermomixer. It is important to perform the following steps precisely to guarantee efficient annealing.
a) Heat to 95 °C for 2 min with shaking at 1100RPM.
b) While shaking, manually decrease the temperature by 2°C every minute, until you reach 50°C.
c)
Stop the shaking but do not take the samples out or
turn off the heating. Move to step 89.
CRITICAL Do not put the thermomixer's cover over the samples during the annealing since this would not allow for an even drop of temperature.
5) Thermostable RNase H Digestion. Timing 1h
89. Without taking the samples out of the thermomixer, add the following components to the experimental or control samples in the specified order (MgCl2 first):
|
EXPERIMENTAL (+RNASE H) DIGESTION REACTION |
VOLUME |
|
Hybridized RNA |
+116 µl |
|
MgCl2 (55mM stock solution)* |
2 µl |
|
RNase H Reaction Buffer (10X) |
20 µl |
|
Thermostable RNase H |
4 µl |
|
Nuclease-free Water |
to 200 µl |
|
Total volume |
200 µl |
|
CONTROL (-RNASE H) REACTION |
VOLUME |
|
Hybridized RNA |
+116 µl |
|
MgCl2 (55mM stock solution)* |
2 µl |
|
RNase H Reaction Buffer (10X) |
20 µl |
|
Nuclease-free Water |
To 200 µl |
|
Total volume |
200 µl |
* MgCl2 is added to neutralise the EDTA in the hybridization buffer. This is essential for the activity of the RNase H enzyme. If for, whatever reason, the final concentration of EDTA is modified, you must insure that the amount of added MgCl2 is adjusted accordingly.
90. Close the caps and turn on the shaking (1100RPM)
91. Incubate the tubes with shaking for 1 h at 50°C.
92. Stop the thermomixer and take the
samples off.
93. Centrifuge samples at 12000g for 3 min.
94. Transfer the supernatant into a new
tube.
CRITICAL Make sure to not transfer any insoluble particles collected at the bottom.
95. Take 1/10 of the sample for analysis of RNA degradation by RT-qPCR or RNA-sequencing (See Appendix). You can snap freeze and store this sample for up to 3 months at -80°C if needed.
96. Proceed with the remaining 9/10th
of the sample to step 97.
CRITICAL Do not stop at this step with the protein preparation.
6) RBPs extraction. Timing 20h
97. Add 900 µl of TRIzol™ LS to each sample and vortex.
98. Add 200 µl of chloroform and vortex the sample until the content becomes homogeneous.
99. Centrifuge for 15 min at 12,000
g at 4°C. Sample should phase separate into the three aqueous, interface, and
organic phases.
CRITICAL It is
important to check that an interface still forms after the phase separation.
Lack of a visible interface would indicate non-specific degradation of protein-bound
RNAs.
100. Recover the releases RBPs by pipetting the organic phase into a new tube.
CRITICAL Only recover the organic phase, free of any interface. If pipetting all of the organic phase while excluding the interface is difficult, some of the organic phase may be left behind to insure no interface carry over.
101. Add 100% cold acetone for a final concentration of 80% acetone,
in order to precipitate the released RBPs. For example, if 300 µl of the
organic phase is collected, add 1200 µl of 100% acetone. Vortex the mix
thoroughly.
PAUSE POINT Precipitate the released RBPs in acetone by incubating at -20°C overnight.
102. Centrifuge for 20 min at 16,000 g at 4°C.
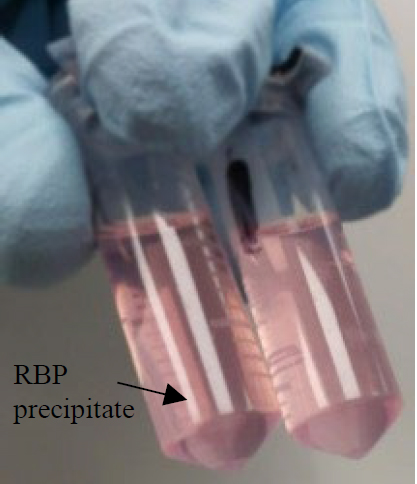
103. Discard the supernatant. RBPs should have precipitated and therefore be present as a pellet after centrifugation (as shown in the image).
Note: Depending on the RNA target and amount of input material, the pellet may be large or very small.
104. Add 1 ml of 80% acetone to wash the pellet.
105. Vortex and centrifuge for 15 min at 16,000 g at 4°C.
106. Discard the supernatant and repeat step 104 to 105.
107. Discard supernatant and air-dry the pellet for 5 min.
7) Sample preparation for shotgun proteomics. Timing 20h
108. Recover RBP
pellet in 100 µl 8M Urea in ABC buffer.
CRITICAL Prepare a fresh urea solution on the day.
CRITICAL Pipette the sample up and down thoroughly to
resuspend pellet as much as possible. Any remaining insoluble material should
dissolve over the course of the trypsin digestion (step 112).
109. Add DTT from a 1M stock to reach a final concentration of 10 mM DTT. Incubate the samples for 30 min at room temperature (20–25 °C) to reduce the proteins.
110.
Add IAA from a 0.55M stock to reach a final concentration of 55 mM. Incubate
the samples for 30 min at room temperature (20–25 °C) in the dark to alkylate
the reduced residues.
CRITICAL IAA is unstable and light-sensitive. Prepare the solution
immediately before use and ensure alkylation is performed in the dark.
111. Dilute the Urea concentration to 2M by adding ABC buffer to the samples.
112. Add 1µg Trypsin per sample and incubate samples at room temperature (20–25 °C) for 16 h.
113. Add 1 volume of Stop 4 buffer to the digested samples.
114. Proceed with C18-based clean up of the samples (e.g. stage tips) to remove salts.
115. Inject the purified peptides into LC-MS/MS (procedure is variable depending on the MS instrument used).
116. Search and quantify the identified RBPs using Label-free Quantification (LFQ). Multiple search and quantification computational pipelines can be used. We routinely use Maxquant and Perseus software packages for the data analysis steps.
Appendix: RT-qPCR and RNA-seq analysis of RNase H digestion efficiency
and specificity
To retrospectively analyse the efficiency and specificity of RNase H mediated target degradation, the 1/10th aliquots taken after RNase H treatment can be used to purify RNA and analyse this in a targeted (RT-qPCR) or untargeted (RNA-seq) manner. For this purpose, follow the below steps:
- Add 110 µl of proteinase K buffer to each taken aliquot.
- Digest the proteins by adding 10 µl Proteinase K and incubating for 1 h at 25 °C.
- Perform an additional Phase separation by adding TRIzol™ LS to the Proteinase K reaction. Collect the free RNA (upper phase). Purify the RNA according to the TRIzol™ LS manual.
- Perform RNA quantification using Qubit.
PAUSE POINT Store extracted RNA for up to 3 months at -80 °C if needed.
- Perform RT-qPCR quantification of the target as well as independent control transcripts to assess degradation efficiency. Alternatively, the purified RNA can be subjected to whole-transcriptome RNA-seq to assess RNAse H-mediated degradation efficiency and specificity in a global unbiased manner.
